
Computer Systems Validation (CSV) acts as the main regulatory control framework for guaranteeing that computerized systems utilized within GxP-regulated life science industries such as the pharmaceutical, biotech, and medical device industries function in a consistent, reliable manner that fully complies with internationally accepted quality expectations. It is a special process aimed at creating the necessary documentation that confirms a particular computer system operates precisely as planned. In today's highly competitive world of global production, the magnitude of this process is enormous, with the global CSV market predicted to reach USD 7.33 billion in 2032.
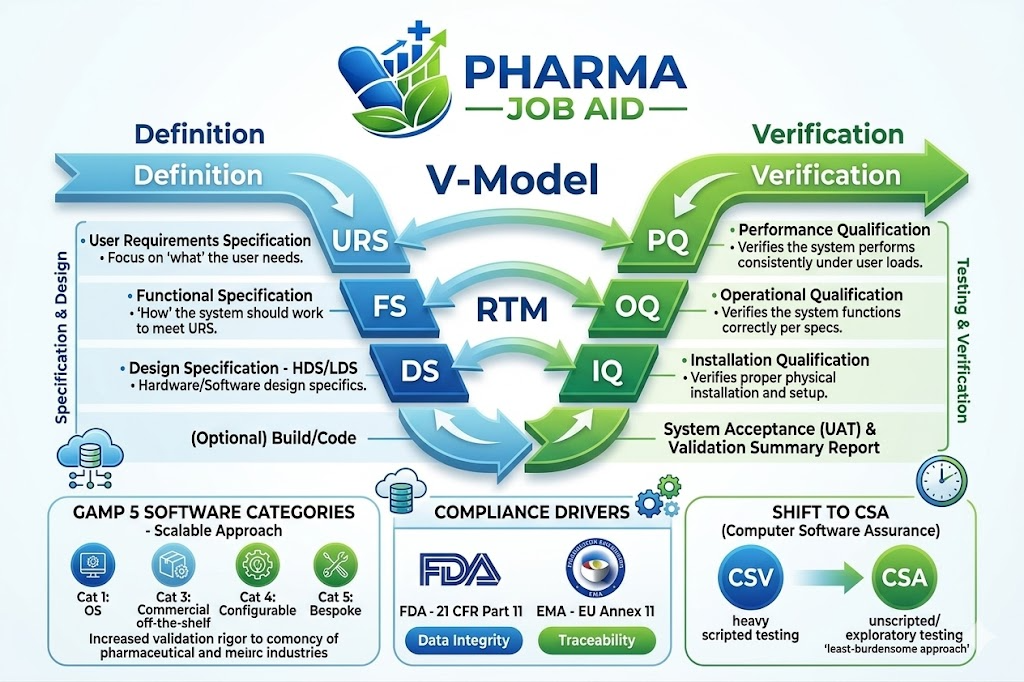
Until recently, the main structural principle for such an approach was the V-Model – a method that involves developing a connection between development requirements and verification requirements in a visual and conceptual way. However, the effectiveness of the systems validation process has proven its ability to impact operations in a positive way. Indeed, the establishment of validated software systems has led to the reduction of average reporting time for clinical adverse events from 16.2 to only 2.3 days, thus achieving an operational improvement of 86%. In today’s world, software systems are extremely complex and characterized by an incredibly large number of decision-making points, complex database interactions, multi-level abstractions, and regular changes. Thus, the evolution of the life sciences industry calls for a significant shift from traditional and documentation-driven linear V-Models to flexible and risk-based Computer Software Assurance (CSA) approaches.
V-Model Structure and V-Model Operation
Understanding the V-Model requires the placement of the model in the context of the computerised system lifecycle structure. In general, there are four main sequential phases to a system lifecycle, namely the concept phase, project phase, operational phase, and retirement phase. In the concept phase, the regulated user prepares a general description of the system, selecting the potential vendors for the software/hardware, determining cost estimates, and developing a basic business justification. The project phase involves designing, configuring, and qualifying the system. Following the successful completion of the project phase, the operational phase deals with keeping the system in a controlled documentation condition via security oversight and monitoring as well as managed change activities. Finally, in the retirement phase, the software and hardware are decommissioned, and secure archiving of data and retrievable records is achieved.
In addition to the aforementioned phases of the computerised system lifecycle, several concurrent support activities should be performed such as risk management, document control, security administration, and correction repair work. The V-Model itself becomes the structural methodology used during the project phase to transform user needs into technical requirements.
The structure of the V-Model includes two arms – the descending specification arm and ascending verification arm. The descending arm of the V-Model begins with the User Requirement Specification (URS). This document is written in plain language to describe business needs without getting into technical detail. From URS comes the development of the Functional Specification (FS), which specifies how the system should operate to satisfy the needs described in the URS. For custom code and custom physical layout development, a Design Specification (DS) document is prepared, which becomes a blueprint showing architecture, data flows, API interfacing, system configurations, etc. Finally, at the vertex of the V, the physical system configuration and software code is generated.
As noted earlier, the ascending arm of the V-Model constitutes the process of verifying specifications laid out along the descending arm. The first verification gate is the installation qualification (IQ), which corresponds to design specification. The installation qualification confirms proper software installation, database tables installation, and correct network architecture based on the vendor specifications. It does not qualify application business logic but verifies version control, directories, Active Directory, and database connection strings.
Further on the ascending arm we see the Operational Qualification (OQ), which corresponds to the FS. The OQ verifies that all individual functionalities described:
| V-Model Specification (Left Arm) | Corresponding Verification (Right Arm) | Core Technical and Operational Verification Objectives | Primary Executing Parties |
| User Requirements Specification (URS) | Performance Qualification (PQ) | Verifies the system performs reliably when executing end-to-end business workflows under actual production conditions, including high database volumes, load capacities, and connected system integrations. | Regulated End Users, Business Process Owners, Quality Assurance (QA) |
| Functional Specification (FS / FRS) | Operational Qualification (OQ) | Verifies the software executes individual functional parameters, boundary limits, negative testing conditions, security access controls, and electronic signature trails. | Validation Specialists, System Engineers, Quality Assurance |
| Design Specification (DS) | Installation Qualification (IQ) | Verifies the software code, database tables, network protocols, server instances, and hardware components are correctly installed and version-controlled per developer blueprints. | IT Infrastructure Engineers, Database Administrators, System Integrators |
GAMP 5 Categorization of Software and Proportionate Validation Scaling
To reduce unnecessary administrative burden and allocate limited resources where they are most needed, the ISPE GAMP 5 guidelines use a systematic approach to software classification. Published in 2008 and updated to GAMP 5, second edition, in July 2022, the guidelines adopt a risk management approach, which scales validation efforts in proportion to system complexity, innovation, and software category.
An important change in the second edition of the guidelines is that software categories must not be seen as an isolated set of checklists but rather as a continuous spectrum. In contemporary life science operations, computerised systems are virtually always made up of composite architectures. As an example, a heavily configured cloud application (Software Category 4) runs on virtualized server and database environments (Software Category 1). Hence, organisations are supposed to think critically, as well as consider other variables such as the criticality of the system and capabilities of vendors, among others.
Classification system for GAMP 5 software is described below:
Category 1: Infrastructure Software: Includes basic software platforms such as operating systems, database management systems, hypervisors, and middleware. They provide hosting environment for other software systems that cannot be customized by the users. Software systems under category 1 carry minimal risks; therefore, they are qualified through documentation of software, version, and location along with stringent change control procedures. Interestingly, office tools such as Microsoft Excel are classified as Category 1 unless they involve building customized data tracking spreadsheets in which case they fall into category 5.
Category 2: Firmware (Retired): Formerly known as the category used to define embedded software logic in GAMP 4, the use of this category has been discontinued since the inception of GAMP 5. Embedding systems are much more sophisticated today. Therefore, firmware is currently defined under Category 3, 4, or 5 based on the software sophistication and GxP impact.
Category 3: Non-configured Products: Includes COTS applications that are used directly ‘as-is’ in their initial installations without any additional configurations aside from their default settings. The validation process for this category involves checking whether the application has been properly installed (Installation Qualification) and performing minimum, risk-oriented operations qualification test on the software.
Category 4: Configured Products: Includes configurable software such as LIMS, MES, SCADA, ERP, and Building Management Systems (BMS), which involve customization through business rules, scripting, and other configurations without changing the source code of the software. In this category, the ‘configuration’ refers specifically to assigning values to variables present in the software. Category 4 software validation requires specification of software configuration requirements, thorough supply quality audits, and functional testing of customized business rules.
Category 5: Custom Applications: Includes all custom software developed in-house or externally and delivered as bespoke solutions to specific business problems. Such software includes customized add-ons and databases and standard spreadsheets having complex custom VBA macros. The biggest concern with category 5 is the presence of bugs within the software due to complex coding. Therefore, a detailed software development life cycle validation approach is required in such cases.
- GAMP 5 Category
- Relative Risk
- Core Operational Definition
- Scaling and Validation deliverables
- Representative Industry Examples
Category 1: Infrastructure Software
Low
Generic, standardized platform systems that establish hosting environments for GxP business applications.
Documentation of software name, version, and location; hardware/virtual server platform qualification; change control.
Windows Server, Linux OS, Oracle DBMS, SQL Server, Active Directory, VMware.
Category 3: Nonconfigured Products
Moderate-Low
Standard commercial software used in its default configuration without customization.
URS; confirmation of intended use; installation verification; limited operational testing of default functions.
Standard laboratory instruments’ embedded software, off-the-shelf analytical packages.
Category 4: Configured Products
Moderate
Commercial software packages modified via business rules, configurations, and user parameters without changing source code.
Full URS; functional and configuration specifications; supplier audits; risk-based testing of configured workflows.
Veeva Vault QMS, SAP ERP, LabWare LIMS, Emerson Syncade MES, Building Management Systems (BMS).
Category 5: Custom Applications
High
Bespoke software developed to address proprietary, unique organizational processes.
Complete design specification; mandatory source code reviews; developer unit testing; full IQ, OQ, and PQ validation.
Custom ERP modules, proprietary real-time batch release engines, spreadsheets containing custom VBA macros.
Global Regulatory Frameworks: FDA 21 CFR Part 11 and EU GMP Annex 11
Computerized Systems Validation is a worldwide regulatory requirement. However, these regulatory bodies adopt different approaches towards such requirements. The primary one in the U.S. is FDA 21 CFR Part 11 regulations, defining when electronic records and signatures become trustworthy, secure, and equivalent to paper records and handwritten signatures. As for Europe, the principle guideline is the EU GMP Annex 11 embedded in the overall EU GMP guidelines.
The key difference is that FDA 21 CFR Part 11 regulation is an enforceable document focusing on electronic records and signatures, whereas EU GMP Annex 11 is a more general system-related guideline covering the whole cycle of computerized systems used in medicinal manufacturing within the scope of GMP requirements.
Both frameworks receive regular updates. In particular, the European Commission has issued a major draft revision of Annex 11 alongside Chapter 4 (Documentation) and an entirely new Annex 22 (Artificial Intelligence) in July 2025. This revision, finishing its public consultations in October 2025 and expected to be completed by mid-2026, codifies data integrity principles of ALCOA++ in EU GMP. Moreover, it imposes extremely strict validation requirements for AI/ML systems applied in pharmaceuticals.
Compliance Attribute
FDA 21 CFR Part 11 (United States)
EU GMP Annex 11 (European Union)
Regulatory Scope
Focuses specifically on the trustworthiness, security, and attributability of electronic records and electronic signatures.
Governs the broader lifecycle management of computerized systems operating within GxP manufacturing environments.
Validation Context
Requires system validation to ensure electronic records are accurate, reliable, and consistently perform to prevent data manipulation.
Mandates comprehensive validation across all stages of the system lifecycle, from URS to design, testing, operation, and retirement.
Risk Management
Implicitly expected; however, specific risk-based categorization frameworks are not explicitly outlined.
Explicitly mandates documented risk management across all lifecycle stages to determine the scale of validation and controls.
Supplier Oversight
Allows the use of vendor documentation but places the primary compliance burden on the regulated company.
Formally mandates supplier qualification, vendor quality audits, and clear, written quality agreements between IT and suppliers.
Periodic Review
Not explicitly dictated within Part 11; typically driven by cGMP predicate rules.
Formally mandates periodic reviews of system performance, audit trails, security access, incident history, and validation status.
Operational Control
Requires specific written procedures for system access, record retention, change control, and training records.
Demands clear allocation of roles, defining responsibilities for System Owners, Process Owners, and IT support teams.
Common Regulatory Vulnerabilities and Audit Findings
From the examination of the warning letters on FDA Form 483 violations, the main causes of the failure to implement the V-model, as well as data integrity control problems, are evident to be the main causes of computer system deficiency in most cases. The major causes include the past practices, mistakes in paper-based documentations, and ineffective analysis of logs of the system.
- Underlying Compliance Vulnerability
- Technical and Operational Remediation Strategy
- Inadequate Validation of Spreadsheets
- Critical calculations and data entries that drive batch release decisions are managed on unvalidated, unsecured Excel spreadsheets. Lock cell formulas under document control, version-control the file structure, and restrict system-level edit access.
- Missing or Retroactive Traceability
- Validation test cases cannot be mapped back to specific URS requirements, or the RTM is compiled after testing has completed. Maintain a live RTM from day one of the project, linking every specification to an executed test script with automated tools.
- Disabled or Unreviewed Audit Trails
- Audit trail configurations are disabled, or there is no documented procedure for the routine, GxP-relevant review of system logs. Configure automated, time-stamped, tamper-evident audit trails that record user name, action, timestamp, old value, and new value.
- Shared and Default User Accounts
- Multiple system users log in under a generic “Administrator” or “Operator” account, destroying data attribution and traceability.
- Implement role-based User Access Management (UAM) with unique, individual accounts, single sign-on (SSO), and MFA.
- Undocumented Deviation Logs
- Testing failures or system anomalies discovered during OQ/PQ execution are resolved “on-the-fly” without being formally logged.
- Implement a mandatory, pre-approved deviation logging protocol requiring formal QA review, impact assessment, and CAPA resolution.
- Lack of Data Retention & Archiving
- No documented plan or verified procedure for database backup, disaster recovery testing, or long-term secure archival.
- Implement regular, automated backups, test system restore protocols routinely, and use data checksums to verify archive integrity.
The Rise of Computer Software Assurance (CSA) as a Contemporary Trend
For a long time, Computer System Validation (CSV) has been regarded as creating an environment of paperwork focused on regulatory compliance. The reality is that validation experts spend up to 80% of their time working out complicated test scenarios and generating repetitive screen shots, leaving about 20% for testing key features of the computer systems. Addressing this issue, the US Food and Drug Administration published a draft version of the final guidance on “Computer Software Assurance (CSA) for Production and Quality System Software” dated September 24, 2025. Another version was published on February 3, 2026.
It should be stressed that Computer Software Assurance does not supersede any regulations or laws. On the contrary, it enhances the existing validation process through promoting critical thinking, scientific risk analysis, patient safety, and effective testing methods. It should be noted that CSA is different from Software as a Medical Device (SaMD) and Software in a Medical Device (SiMD). This approach can be applied to production and quality systems software including Laboratory Information Management Systems (LIMS), Manufacturing Execution Systems (MES), Enterprise Resource Planning (ERP), Computerized Maintenance Management Systems (CMMS), Learning Management Systems (LMS), and eQMS.
The core lifecycle process of CSA involves six essential steps described as follows:
Determination of Intended Use: Determine whether the software’s capabilities will support the production process as well as the quality system based on the software’s capability.
Determination of Risk-Based Approach: Categorize each individual software function/feature/operation into either High Process Risk (failure poses quality/patient safety concern) or Not High Process Risk.
Software Change Management: Use the risk-based approach in the management of changes made to the software post-production through system updates and patches.
Determination of Appropriate Assurance Activities: Select testing activities appropriate to the level of process risk.
Additional Mitigation Strategy: Use existing process control strategies, redundancy and cybersecurity mechanisms to reduce the burden of validation.
Establishment of an Appropriate Record: Collect objective evidence indicating that the system is performing as intended. The FDA recommends objective evidence generated by the system (system logs, database transactions, etc.) as opposed to manually captured screens.
Additionally, CSA formally specifies three testing methods in order to facilitate validation according to the process risks involved:
Robust Scripted Testing: Test method applied for high process risks where test actions are clearly defined with expected results, documented objective evidence collected, and traceable to the user requirements.
Limited Scripted Testing: Intermediate test strategy that involves the application of robust script testing for high process risk functions and unscripted testing for low and medium process risks.
Unscripted Testing: Test strategy used for low process risk functions that involves testing the software in action without using any step-by-step instructions for testing. Unscripted testing can be subcategorized as follows:
Ad-hoc Testing: Specification-based testing performed by executing specified instructions without detailed written instructions.
Error Guessing Testing: Testing based on tester’s experience and knowledge of the software and errors observed in past applications of similar software.
Exploratory Testing: Heuristic experience-based testing based on tester’s experience/knowledge.
Validation Parameter
Traditional Computer System Validation (CSV)
Modern Computer Software Assurance (CSA)
Core Testing Focus
Verifies every single software requirement uniformly, regardless of risk or system complexity.
Focuses validation efforts heavily on high-risk system features that directly impact product quality and patient safety.
Testing Deliverables
Relies on manual, step-by-step scripted testing for 100% of the defined requirements.
Employs a risk-proportionate mix of robust scripted, limited scripted, and unscripted (exploratory, ad-hoc) testing.
Evidence Collection
Captures manual, paper-heavy documentation and verbose screenshots for every executed step.
Focuses on lean documentation, utilizing automated system logs, digital audit trails, and automated testing metadata.
Vendor Test Strategy
Often repeats vendor qualifications internally, resulting in redundant validation cycles.
Leverages quality assessments and test artifacts from qualified, audited vendors to minimize local duplication.
Lifecycle Methodology
Primarily aligned with linear, waterfall-like development lifecycles.
Aligns naturally with agile development, continuous deployment (CI/CD) pipelines, and SaaS platforms.
Agile Life Cycles and Non-Linear V-Model
The development of software has evolved beyond traditional linear or waterfall-type approaches. In the life sciences domain, it was traditionally thought that the V-Model must be conducted in one static macro-cycle fashion. The legacy process entailed defining all the requirements upfront, conducting configurations separately, and testing only towards the end, resulting in long deployment times.
GAMP 5 Second Edition (2022) posits that system lifecycles are not necessarily linear. Modern systems engineering practice favors incorporating agile, iterative, and incremental approaches in order to achieve ROI optimization and maintain innovation. Under an agile model, the V-Model becomes a series of “micro-V-sprints.”
Here, user stories (micro-URS entries) and functional acceptance criteria (micro-FS entries) are managed using unified specifications, as described in GAMP 5 Appendix D1 (Specifying Requirements). In lieu of running a massive V-Model lifecycle, a separate lifecycle design, configuration, and automated testing cycle runs during each sprint.
To remain agile, GAMP 5 Appendix D5 (Testing of Computerized Systems) highlights the use of automated testing tools and modern digital traceability capabilities. Automated testing solutions support continuous regression testing to ensure that system behavior is correct with each new feature deployment.
Additionally, GAMP 5 Appendix M11 (IT Infrastructure) discusses SaaS continuous patching cycles. It allows QA and IT teams to negotiate formal agreements that clarify which are considered low-risk changes such as operating system security updates, which then avoid following a cumbersome change control process. Critical thinking makes it possible to skip installation verification steps when installation confirmation is achieved through functional testing.
Recommendations for Strategic CSA Validation
For leveraging new generation validations and being prepared for upcoming inspections, companies in the life science sector must consider implementing the following steps:
1. Implement a Risk-Based CSA Process
Establish official criteria by which features of computer software can be categorized as having a “High Process Risk” or not having a high process risk. In other words, this step involves assessing the specific process that is supported by the software to determine whether a problem with the software may affect product quality, data integrity, and patient safety.
2. Move from Paper-based Documentation to Digital
Revise standard operating procedures in such a way that there is a switch from paper-based screenshots and signatures to automated logging, audit trails, and electronic signing systems. Also, use validation software to maintain the traceability matrix dynamically rather than retroactively.
3. Implement a Supplier Qualification and Utilization Program
Implement an evaluation program to qualify suppliers, in particular vendors of SaaS and platforms used for critical processes. Should a vendor have an existing validated software pipeline, use it within the local validation master plan to avoid unnecessary IQ/OQ testing.
4. Conduct a Pilot CSA Project
Before transitioning into digital, implement a targeted CSA pilot project in which the organization selects a software system (for example, LMS or BI reporting software) that will support quality but not production to practice risk-based testing and exploratory testing and generate documentation.
- pharmuni.com What is Computer System Validation (CSV)? – Pharmuni Opens in a new window
- gmpinsiders.com GAMP 5 In CSV: Definition, Categories, And Pharma Guidelines …Opens in a new window
- validatorvlms.com Computer System Validation Automation | Validator Compliance Solutions – Validator VLMS mOpens in a new window
- avslifesciences.com 7 Key Insights on CSV Full Form in Pharma Compliance – AVS Life Sciences Opens in a new window
- ispewebassets.org Computer Systems Validation: A Systems Engineering Approach Opens in a new window
- gmpinsiders.com CSV Vs CSA: Key Differences In Software Validation | GMP Insiders Opens in a new window
- pscsoftware.com GAMP 5 Second Edition Explained: Key Changes and Updates – PSC Software Opens in a new window
- getreskilled.com What is Computer System Validation (CSV) in Pharma? – Get Reskilled Opens in a new window
- perfval.com 10 Strategic Tips for Computer System Validation in the Pharmaceutical Industry Opens in a new window
- therxcloud.com Computer System Validation Flow Chart: FDA Compliance Guide – RxCloud Opens in a new window
- govalidation.com IQ OQ PQ Validation 2026: Complete Guide for Pharma & Life … Opens in a new window
- vmtspharmasoftware.com Traceability Matrix in CSV Pharma Industry: Why It’s Non-Negotiable – Opens in a new window
- gmpinsiders.com Annex 11 Vs 21 CFR Part 11: Comparison And GMP Requirements Opens in a new window
- intuitionlabs.ai GAMP 5 Categories Explained: Software, Risk & Examples …Opens in a new window
- intuitionlabs.ai GAMP 5 Categories Explained: Software, Risk & Examples – IntuitionLabs Opens in a new window
- blog.montrium.com GAMP® 5 Second Edition: What you should know – Montrium Blog Opens in a new window
- qbdgroup.com GAMP categories for computerized systems: what… | QbD Group Opens in a new window
- sware.com What is GAMP 5 in Pharma? An Essential Resource for Compliance and Validation – Sware Opens in a new window
- intuitionlabs.ai Computer System Validation in Pharma & Biotech Compliance … Opens in a new window
- simplerqms.com FDA 21 CFR Part 11 vs. EU Annex 11: Key Requirements and …Opens in a new window
- sgsystemsglobal.com EU GMP Annex 11 – Computerised Systems & Data Integrity Opens in a new window
- pscsoftware.com FDA CSA Guidance | Risk-Based Validation in Pharma – PSC Software Opens in a new window
- blog.pqegroup.com Toward a Least-Burdensome Approach to Computer Software … Opens in a new window
- mastercontrol.com CSA vs. CSV: FDA Computer Software Assurance Draft Guidance Explained – MasterControl Opens in a new window
- pharmanow.live Computer System Validation (CSV) vs Computer Software Assurance (CSA) – Pharma Now Opens in a new window
- bluemountain.io FDA CSA Guidance: Risk-Based Software Validation for GMP – Blue Mountain Opens in a new window
- scilife.io CSV vs. CSA: What Are the Main Differences? – Scilife Opens in a new window
- rookqs.com FDA Computer Software Assurance 2025 Guidance – Rook Quality Systems Opens in a new window
- ddismart.com FDA: Software Assurance guidance for Production and Quality System Software – – DDi Opens in a new window
- spectroscopyonline.com CSA: Much Ado About Nothing? – Spectroscopy Online Opens in a new window
- qbdgroup.com GAMP 5 guide 2nd edition: what’s new? | QbD Group Opens in a new window
- intuitionlabs.ai GAMP 5 Second Edition: A Guide to Key Changes & Updates | IntuitionLabs