
Introduction
Analytical Development is a core scientific discipline within pharmaceutical research and development that ensures a drug product consistently meets its predefined quality attributes. It involves the development, validation, and lifecycle management of analytical methods for assessing identity, purity, potency, and performance. These analytical procedures are fundamental to regulatory submissions. They must comply with global standards set by organisations such as the International Council for Harmonisation, World Health Organisation, and U.S. Food and Drug Administration.
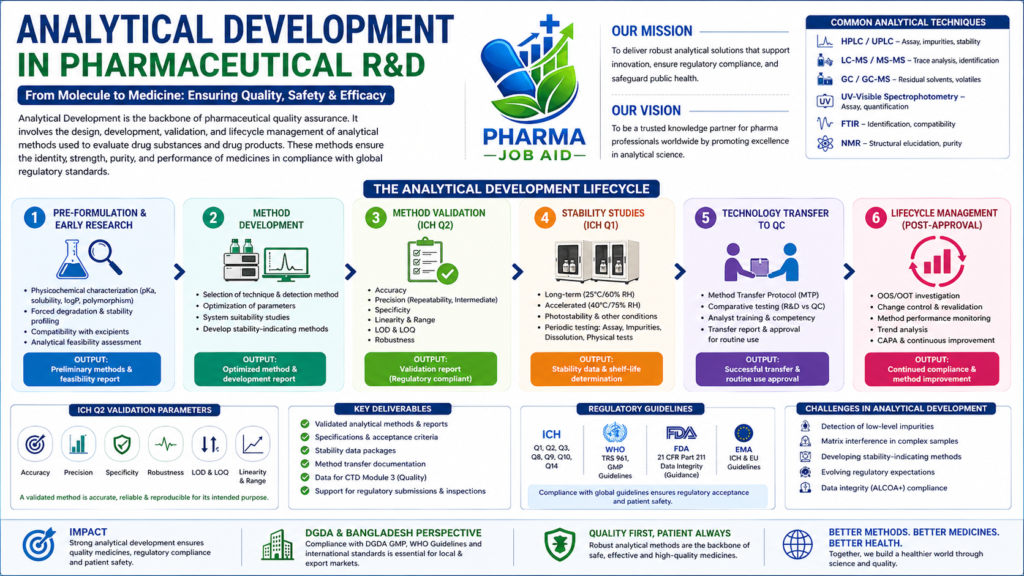
Role of Analytical Development in Drug Lifecycle
Analytical development supports every stage of the drug lifecycle, beginning from early discovery through commercial manufacturing and post-marketing surveillance. During the initial phases, it helps characterise the API and supports formulation design. As development progresses, it ensures that analytical methods are robust and capable of detecting impurities, degradation products, and variability in manufacturing. In the commercialization stage, these methods are transferred to quality control laboratories for routine testing, ensuring batch-to-batch consistency and regulatory compliance.
Preformulation and Characterisation Studies
In the early stage of R&D, analytical scientists perform extensive characterisation of the drug substance. This includes studying physicochemical properties such as solubility, pKa, polymorphism, and thermal behaviour. Stress testing under conditions like heat, light, humidity, and oxidation is conducted to understand degradation pathways and to develop stability-indicating methods. These studies are crucial for selecting suitable excipients and designing stable formulations, while also providing the scientific basis for future analytical method development.
Analytical Method Development
Analytical method development is a systematic process where scientists design and optimize procedures for testing drug substances and products. Techniques such as High-Performance Liquid Chromatography (HPLC), Gas Chromatography (GC), Liquid Chromatography–Mass Spectrometry (LC-MS), and UV-Visible spectroscopy are commonly employed. The goal is to develop methods that are accurate, precise, specific, and reproducible. Parameters like mobile phase composition, column selection, flow rate, and detection wavelength are carefully optimized to achieve reliable separation and quantification of analytes, including impurities and degradation products.
Method Validation (ICH Q2 Compliance)
Once developed, analytical methods must be validated to demonstrate their suitability for intended use. Validation is conducted in accordance with ICH Q2 guidelines and includes parameters such as accuracy, precision, specificity, linearity, range, robustness, limit of detection (LOD), and limit of quantitation (LOQ). Validation ensures that the method consistently produces reliable results under defined conditions. The outcome of this process is a comprehensive validation report, which becomes a critical part of regulatory documentation.
Stability Studies and Shelf-Life Determination
Analytical development plays a key role in stability studies, which are conducted to determine the shelf life of a pharmaceutical product. These studies follow ICH Q1 guidelines and involve storing samples under various environmental conditions such as long-term (25°C/60% RH) and accelerated (40°C/75% RH). Analytical testing is performed at predefined intervals to monitor changes in assay, impurity levels, dissolution, and physical characteristics. The data generated helps establish expiry dates, storage conditions, and packaging requirements.
Technology Transfer to Quality Control
After successful development and validation, analytical methods are transferred from R&D to the Quality Control (QC) laboratory. This process, known as method transfer, ensures that the receiving laboratory can perform the method with equivalent accuracy and precision. It involves preparing a method transfer protocol, conducting comparative testing, and training analysts. Successful transfer is documented through a method transfer report, enabling routine quality testing in compliance with GMP requirements.
Lifecycle Management and Continuous Improvement
Analytical methods are not static; they require ongoing monitoring and improvement throughout the product lifecycle. Post-approval activities include handling Out-of-Specification (OOS) and Out-of-Trend (OOT) results, implementing change controls, and performing revalidation when necessary. Continuous trend analysis and integration with CAPA systems ensure that analytical performance remains consistent and compliant with regulatory expectations, especially in a GMP-regulated environment.
Regulatory Significance and Compliance
Analytical development data form a major part of regulatory submissions, particularly in CTD Module 3 (Quality). Regulatory authorities such as the European Medicines Agency and U.S. Food and Drug Administration rigorously evaluate analytical methods, validation data, and stability results before granting product approval. Compliance with international guidelines ensures global acceptance and facilitates market authorization in multiple regions.
Challenges in Analytical Development
Analytical development faces several practical and scientific challenges, including the detection of low-level impurities, matrix interference in complex formulations, and the development of stability-indicating methods. Additionally, maintaining data integrity in line with ALCOA+ principles and adapting to evolving regulatory expectations require strong quality systems and skilled personnel. Addressing these challenges is essential for ensuring robust and reliable analytical performance.
Strategic Importance in Modern Pharma
Analytical Development (AD) is not just a laboratory activity—it is a regulatory-critical, risk-based scientific system that underpins product quality throughout the drug lifecycle. It ensures that every batch released into the market consistently meets predefined Critical Quality Attributes (CQAs) such as assay, impurities, dissolution, and content uniformity. Modern analytical development is deeply integrated with Quality by Design (QbD) principles and guided by global frameworks from the International Council for Harmonisation, World Health Organization, and U.S. Food and Drug Administration.
In a DGDA/GMP environment like Bangladesh, analytical development is also a compliance backbone, ensuring audit readiness, data integrity, and international acceptance.
Quality by Design (QbD) in Analytical Development
Modern analytical development follows QbD, where methods are not just developed but scientifically understood and controlled.
- Analytical Target Profile (ATP): Defines what the method must measure and its performance criteria
- Critical Method Parameters (CMPs): Variables such as flow rate, pH, column temperature
- Critical Method Attributes (CMAs): Outcomes like resolution, tailing factor, sensitivity
Using statistical tools like Design of Experiments (DoE), scientists define a Method Operable Design Region (MODR)—a multidimensional space where the method consistently performs well. This reduces variability and regulatory risk.
Preformulation & Forced Degradation Studies
Analytical development begins with deep understanding of the API. Forced degradation (stress testing) is a regulatory expectation under ICH.
Typical stress conditions include:
- Acid/Base hydrolysis
- Oxidation (e.g., H₂O₂ exposure)
- Thermal degradation
- Photolysis (UV/light exposure)
These studies help:
- Identify degradation pathways
- Develop stability-indicating methods
- Establish impurity profiles
A well-designed degradation study ensures that analytical methods can separate and quantify all degradation products, which is essential for regulatory approval.
Advanced Analytical Method Development Strategy
Method development is no longer trial-and-error; it is a systematic, risk-based approach.
Chromatographic Development Considerations:
- Column chemistry (C18, C8, Phenyl, etc.)
- Mobile phase (buffer type, pH, organic solvent)
- Gradient vs isocratic elution
- Detection system (UV, PDA, MS)
For Impurity Profiling:
- Resolution between peaks ≥ 2.0 (typical GMP expectation)
- Sensitivity for trace-level impurities (ppm level)
Orthogonal Techniques:
To ensure reliability, multiple techniques are used:
FTIR + NMR (structural confirmation)
HPLC + LC-MS (quantification + identification)
Analytical Target Profile (ATP) and Quality by Design (QbD)
Modern analytical development is increasingly driven by the concept of an Analytical Target Profile (ATP), which defines what the analytical method must achieve in terms of performance characteristics (e.g., accuracy, precision, detection capability). This approach is aligned with Quality by Design (QbD) principles promoted by the International Council for Harmonisation (ICH Q8, Q9, Q14). Instead of relying on trial-and-error, analytical scientists systematically design methods based on risk assessment and scientific understanding. Critical Method Parameters (CMPs) such as pH, column temperature, and mobile phase composition are identified, while Critical Analytical Attributes (CAAs) like resolution and sensitivity are controlled. This structured approach ensures robustness and regulatory flexibility, reducing the likelihood of method failure during routine QC testing.
Forced Degradation and Stability-Indicating Methods
A key requirement in analytical development is the creation of stability-indicating methods, which can accurately detect and separate degradation products from the active ingredient. Forced degradation studies expose the drug substance and product to stress conditions such as acid/base hydrolysis, oxidation, photolysis, and thermal stress. These studies help identify degradation pathways and confirm that the analytical method can specifically quantify the API without interference. Regulatory agencies including the U.S. Food and Drug Administration and World Health Organization expect well-documented degradation profiles as part of submission dossiers, making this step essential for approval.
Impurity Profiling and Control Strategy
Impurity profiling is one of the most critical aspects of analytical development. Impurities may arise from synthesis, degradation, or interaction with excipients. Analytical methods must be sensitive enough to detect impurities at trace levels (often in parts per million). Guidelines such as ICH Q3A and Q3B define acceptable limits and reporting thresholds. Advanced techniques like LC-MS/MS and GC-MS are widely used for identification and quantification. A well-defined impurity control strategy not only ensures patient safety but also supports regulatory acceptance across global markets.
Elemental Impurity and Residual Solvent Analysis
In addition to organic impurities, analytical development must address elemental impurities and residual solvents. Elemental impurities such as heavy metals are controlled as per ICH Q3D, while residual solvents are governed by ICH Q3C. Techniques like Inductively Coupled Plasma Mass Spectrometry (ICP-MS) are used for elemental analysis, while Gas Chromatography (GC) is commonly used for solvent detection. These tests are essential to ensure that toxic substances remain within permissible limits and do not pose health risks to patients.
Dissolution Method Development and IVIVC
For solid oral dosage forms, dissolution testing is a critical analytical tool that evaluates drug release characteristics. Analytical development focuses on designing discriminatory dissolution methods that can detect changes in formulation or manufacturing processes. In advanced cases, an In Vitro–In Vivo Correlation (IVIVC) is established, linking laboratory dissolution data with clinical performance. This reduces the need for extensive bioequivalence studies and accelerates product approval.
Analytical Method Transfer and Verification
Method transfer is a highly regulated process that ensures analytical methods developed in R&D can be successfully executed in QC laboratories or at manufacturing sites. It involves comparative testing, statistical evaluation, and strict documentation. The receiving laboratory must demonstrate equivalent performance through parameters like precision and accuracy. Any variation observed during transfer must be investigated and resolved through CAPA systems. This step is particularly critical for multinational companies transferring technology across different regulatory regions.
Data Integrity and ALCOA+ Principles
In the current regulatory landscape, data integrity is a major focus area. Analytical development must ensure compliance with ALCOA+ principles—data should be Attributable, Legible, Contemporaneous, Original, Accurate, Complete, Consistent, Enduring, and Available. Regulatory bodies such as the U.S. Food and Drug Administration and European Medicines Agency frequently issue warning letters related to poor data integrity practices. Implementation of computerized systems, audit trails, and secure data management practices is essential to maintain compliance.
Use of Advanced Analytical Technologies
With the evolution of pharmaceutical science, analytical development is increasingly adopting advanced technologies. Techniques such as High-Resolution Mass Spectrometry (HRMS), Nuclear Magnetic Resonance (NMR), and Ultra-Performance Liquid Chromatography (UPLC) offer higher sensitivity, speed, and resolution. Additionally, Process Analytical Technology (PAT) enables real-time monitoring of manufacturing processes, supporting continuous manufacturing and real-time release testing (RTRT). These advancements improve efficiency and reduce development timelines.
Role of Analytical Development in Biosimilars and Biologics
Analytical development becomes even more complex in biologics and biosimilars due to the large molecular size and structural complexity of proteins. Techniques such as peptide mapping, electrophoresis, and bioassays are required to evaluate critical quality attributes like glycosylation, aggregation, and higher-order structure. Regulatory agencies demand extensive comparability studies to demonstrate similarity with reference products. Analytical development thus plays a central role in ensuring the safety and efficacy of biologic therapies.
Documentation and Regulatory Filing (CTD Module 3)
All analytical development activities must be thoroughly documented for regulatory submission. This includes method development reports, validation protocols, validation reports, stability data, and specifications. These documents are compiled in CTD Module 3, which is reviewed by agencies such as the European Medicines Agency and U.S. Food and Drug Administration. Clear, scientifically justified documentation increases the likelihood of approval and reduces regulatory queries.
Integration with Quality Systems (QMS)
Analytical development is closely integrated with the pharmaceutical Quality Management System (QMS). Deviations, OOS/OOT results, change controls, and CAPA are all linked to analytical performance. Regular trend analysis helps identify potential issues before they become critical. This integration ensures continuous improvement and compliance with GMP requirements, particularly under DGDA and WHO guidelines.
Conclusion
Analytical Development is the backbone of pharmaceutical quality assurance, providing the scientific evidence needed to ensure that medicines are safe, effective, and consistent. From early-stage characterization to post-marketing lifecycle management, it plays an indispensable role in the success of drug development programs. A well-structured analytical development system not only supports regulatory compliance but also enhances product reliability and patient safety across global markets.