
📄 Document Control & Management System in the Pharmaceutical Industry in Bangladesh: Complete GMP, Compliance & Career Guide
Introduction
In the pharmaceutical industry, documentation is not just a regulatory requirement—it is the foundation of quality assurance, compliance, and patient safety. In Bangladesh, where the pharmaceutical sector supplies nearly 97% of domestic demand and exports to over 150 countries, maintaining a robust Document Control & Management System (DCMS) is essential for sustaining both local and global competitiveness.
Every tablet, capsule, or injectable product manufactured in a pharmaceutical facility is backed by hundreds of documents. These documents serve as proof that each step—from raw material sourcing to final product release—has been performed according to approved procedures.
Without proper document control:
- Products may fail regulatory inspections
- Data integrity can be compromised
- Patient safety may be at risk
This is why global regulatory bodies such as:
- World Health Organization (WHO GMP)
- International Council for Harmonisation (ICH Q7, Q10)
- U.S. Food and Drug Administration (21 CFR Part 211)
require pharmaceutical companies to implement strict documentation systems.
👉 For pharma career resources and jobs, visit: https://pharmajobaid.com/
What is Document Control in Pharmaceutical Industry?
Document control is a structured process that ensures all documents used in pharmaceutical operations are:
- Accurate
- Approved
- Up-to-date
- Traceable
- Accessible
It governs the entire lifecycle of documents including creation, review, approval, distribution, revision, and archival.
Types of Controlled Documents
Pharmaceutical companies manage a wide range of documents such as:
- Standard Operating Procedures (SOPs)
- Batch Manufacturing Records (BMR)
- Batch Packaging Records (BPR)
- Validation Protocols
- Change Control Documents
- Deviation Reports
- Training Records
A proper document control system ensures that:
✔ Only approved documents are used in operations
✔ Obsolete versions are removed immediately
✔ All records are audit-ready and retrievable
Importance of Document Control in Pharma
1. Regulatory Compliance
Pharmaceutical companies in Bangladesh must comply with multiple regulatory frameworks as highlighted in your content , including:
- WHO GMP
- ICH Guidelines (Q7, Q10)
- US FDA (21 CFR Part 211)
- EU GMP
These regulations mandate that all documents must be:
- Prepared
- Reviewed
- Approved
- Controlled
Failure to comply can result in:
- Warning letters
- Product recalls
- Export bans
2. Product Quality & Patient Safety
Documentation ensures consistency in manufacturing processes.
For example:
- If a process is documented correctly, it can be repeated without variation
- Any deviation can be identified and corrected
This ensures that every batch produced meets the same quality standards.
3. Audit & Inspection Readiness
Regulatory authorities such as Bangladesh’s drug regulator:
- Directorate General of Drug Administration
rely heavily on documentation during inspections.
Auditors often say:
👉 “If it’s not documented, it didn’t happen.”
4. Data Integrity (ALCOA+ Principles)
Pharmaceutical documentation must follow ALCOA+ principles mentioned in your file :
- Attributable
- Legible
- Contemporaneous
- Original
- Accurate
- Complete
- Consistent
- Enduring
- Available
These principles ensure that data is reliable and trustworthy.
What is a Pharmaceutical Document Management System (DMS)?
A Document Management System (DMS) in the pharmaceutical industry is not just a storage tool—it is a controlled, validated system that governs the entire lifecycle of GMP-critical documents.
In regulated environments, a DMS must ensure:
- Data integrity (ALCOA+)
- Regulatory compliance
- Audit traceability
- Controlled access
Unlike general document systems, pharma DMS must align with:
- U.S. Food and Drug Administration (21 CFR Part 11)
- International Council for Harmonisation (Q7, Q10)
- World Health Organization GMP
🔍 Core Functions of DMS (Deep Dive)
Your provided content lists the basics — here is the real operational depth:
1. Document Creation (Controlled Authoring)
- Templates are pre-approved by QA
- Authors must follow standard formats
- Metadata included:
- Document ID
- Version number
- Department
- Effective date
🔎 Example:
An SOP for equipment cleaning must include:
- Purpose
- Scope
- Responsibilities
- Procedure steps
- Safety precautions
2. Review & Approval Workflow
A document cannot be used until approved.
Multi-level review includes:
- Department Head → Technical accuracy
- QA → GMP compliance
- Regulatory → Compliance with export requirements
💡 In advanced EDMS:
- Workflow is automated
- Digital signatures are used
- Approval history is stored permanently
3. Secure Storage
Documents must be stored in:
- Controlled physical archives (paper systems)
- Validated servers/cloud (EDMS)
Security measures:
- Role-based access
- Password protection
- Backup systems
4. Retrieval System
A good DMS allows:
- Instant search by document number
- Retrieval during audits within minutes
⚠️ During inspections, inability to retrieve documents quickly is a major compliance failure.
5. Version Control (Critical Function)
Version control ensures:
- Only latest version is in use
- Old versions are archived
Example:
| Version | Status | Action |
|---|---|---|
| V1.0 | Obsolete | Archived |
| V2.0 | Active | In use |
📊 Types of Document Management Systems (Detailed Comparison)
1. Manual (Paper-Based System)
Still widely used in small Bangladeshi pharma companies.
Advantages:
- Low initial cost
- Simple implementation
Disadvantages:
- No audit trail
- Risk of physical damage
- Slow retrieval
- High human error
📉 Example issue:
A missing BMR page can lead to batch rejection.
2. Electronic Document Management System (EDMS)
Modern, compliant, and globally accepted.
Key capabilities:
- Automated workflows
- Audit trails
- Electronic signatures
- Access control
Compliance:
- 21 CFR Part 11 compliant
- Data integrity ensured
Examples of EDMS software:
- MasterControl
- Veeva Vault
- OpenText
🏗 Structure of Pharmaceutical Documentation System (Expert View)
Your hierarchy is correct — here’s deeper interpretation:
Level 1: Quality Manual
- Describes entire QMS
- Approved by top management
- Rarely changed
📌 Example:
Defines company commitment to GMP and regulatory compliance.
Level 2: Policies
- Broad operational rules
- Not step-by-step
📌 Example:
“Only trained personnel can operate production equipment.”
Level 3: SOPs (Most Critical Level)
- Core of document control
- Used daily
📌 Example SOPs:
- Line clearance SOP
- Deviation handling SOP
- Sampling SOP
Level 4: Records & Forms (Evidence Layer)
These are legal documents.
Examples:
- BMR
- BPR
- Logbooks
- Calibration records
⚠️ These are inspected during audits — errors here can shut down production.
📑 Key Documents in Pharmaceutical Industry (Advanced Insight – Expanded)
In the pharmaceutical industry, documentation is not just a support function—it is a legal, regulatory, and scientific backbone of manufacturing operations. Every document serves as evidence of compliance with Good Manufacturing Practices (GMP) and ensures product quality, traceability, and patient safety.
Below is a detailed breakdown of the most critical pharmaceutical documents:
1. Standard Operating Procedures (SOPs)
SOPs are the foundation of pharmaceutical operations, providing detailed instructions on how to perform tasks consistently and compliantly.
🔍 Key Characteristics of Effective SOPs
- Clear → Written in simple, understandable language
- Stepwise → Logical sequence of actions
- Unambiguous → No room for interpretation
- GMP-Compliant → Aligned with regulatory requirements
- Controlled → Version-controlled and approved
📌 Typical SOP Structure
- Purpose
- Scope
- Responsibilities
- Procedure
- Safety precautions
- Documentation requirements
⚠️ Common SOP Mistakes (Critical for Audits)
- Vague or unclear instructions
- Missing critical process steps
- Lack of revision history
- Outdated procedures still in use
💡 Audit Insight:
Regulators from U.S. Food and Drug Administration often issue observations when SOPs are not followed exactly as written.
2. Batch Manufacturing Record (BMR)
The Batch Manufacturing Record (BMR) is the most important GMP document in pharmaceutical manufacturing.
It provides complete traceability of how a batch was produced.
🔬 Critical Elements of BMR
- Raw material details (name, batch number, quantity)
- Weighing and dispensing records
- Equipment used
- Step-by-step manufacturing process
- In-process quality checks
- Operator signatures and timestamps
💡 Why BMR is Critical
- Acts as legal evidence of manufacturing
- Used during audits and inspections
- Essential for product recall investigations
⚠️ Regulatory Risk:
Any missing or incorrect entry in BMR can result in:
- Batch rejection
- Regulatory warning
- Financial loss
3. Batch Packaging Record (BPR)
The Batch Packaging Record (BPR) ensures that packaging operations are accurate, controlled, and free from mix-ups.
🎯 Key Objectives
- Ensure correct labeling
- Prevent product mix-ups
- Maintain traceability
📦 Includes:
- Packaging line clearance records
- Label issuance and reconciliation
- Packaging materials verification
- Finished product checks
💡 Real Example:
A labeling error (wrong strength printed) can lead to market recall and regulatory penalties.
4. Validation Documents
Validation ensures that processes consistently produce products meeting quality standards.
🔍 Types of Validation
- Process Validation → Confirms manufacturing process reliability
- Cleaning Validation → Ensures no cross-contamination
- Equipment Qualification → Confirms equipment performance
📊 Key Validation Documents
- Validation Protocol
- Validation Report
- Qualification Documents (IQ, OQ, PQ)
💡 Industry Insight:
Validation is mandatory for export compliance to markets like the EU and USA.
5. Change Control Documents
Change control ensures that any modification is evaluated, approved, and documented before implementation.
🔄 Changes Covered
- Equipment changes
- Process modifications
- Raw material changes
- Facility upgrades
📌 Key Steps
- Change request initiated
- Impact assessment
- QA approval
- Implementation
- Effectiveness check
💡 Golden Rule:
👉 “No change without change control approval.”
6. Deviation Reports
Deviation reports document any unexpected event or departure from approved procedures.
⚠️ Examples of Deviations
- Temperature excursion
- Equipment malfunction
- Process deviation
📌 Key Components
- Description of deviation
- Root cause analysis
- Corrective and Preventive Actions (CAPA)
💡 Audit Insight:
Frequent deviations without proper CAPA can lead to regulatory action.
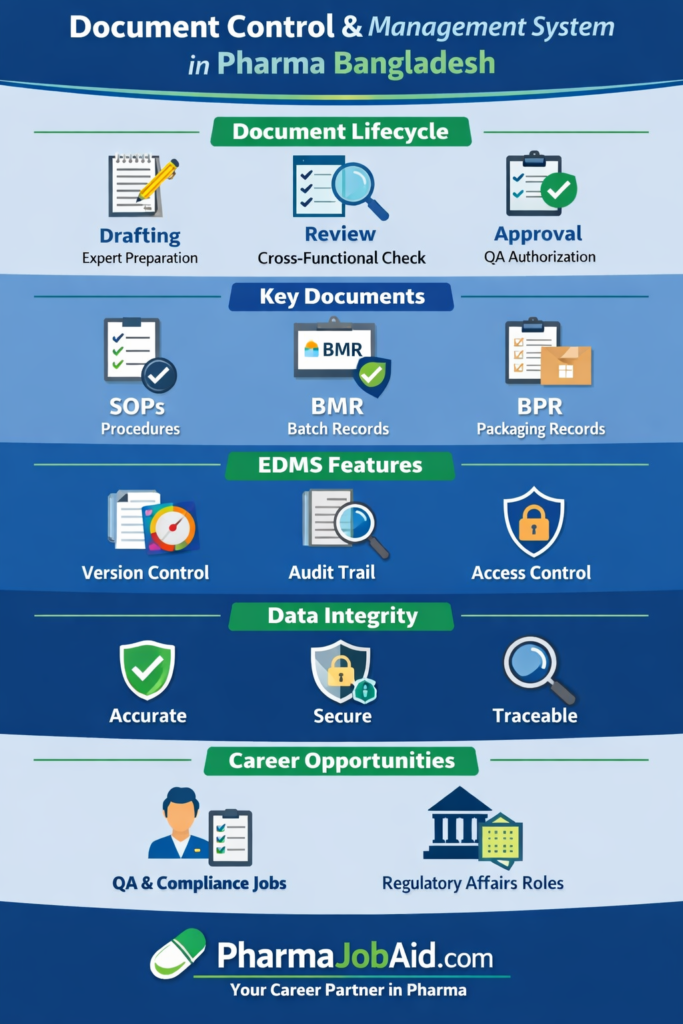
🔄 Document Lifecycle in the Pharmaceutical Industry: Real Industry Practice Explained in Detail
In the pharmaceutical industry, no controlled document can be created and used casually. Every document must pass through a structured document lifecycle to ensure that it is accurate, compliant, traceable, and approved for use. This lifecycle is a core part of the Quality Management System (QMS) and helps companies maintain compliance with GMP, data integrity principles, and regulatory expectations.
A pharmaceutical document lifecycle does not only apply to SOPs. It also applies to quality manuals, batch manufacturing records, validation protocols, specifications, forms, logbooks, and many other GMP documents. Each stage of the lifecycle is designed to reduce risk, prevent errors, and ensure that only the right version of the right document is available to the right people at the right time.
1. Drafting
The first stage of the document lifecycle is drafting. At this stage, the document is created by a person who has the technical knowledge and operational understanding of the process. In most pharmaceutical companies, drafting is usually done by subject matter experts (SMEs) such as QA personnel, production officers, QC analysts, engineering staff, validation teams, or regulatory affairs professionals.
The drafting stage is extremely important because the quality of the final document depends heavily on how accurately the first draft is prepared. A poorly drafted SOP or record format can create confusion, process deviations, training gaps, and compliance risks.
What happens during drafting?
- The need for a new document is identified
- The department prepares the draft using an approved template
- Regulatory and GMP requirements are considered
- Department-specific operational details are added
- The draft is formatted according to company documentation standards
Key drafting considerations
A pharmaceutical document draft should be:
- Clear and easy to understand
- Written in formal and controlled language
- Free from vague wording
- Technically accurate
- Aligned with actual shop-floor practice
- Based on current regulatory expectations
Example
If a company is drafting an SOP for equipment cleaning, the draft must clearly define:
- Which equipment is covered
- Who is responsible for cleaning
- What cleaning materials are used
- How the cleaning is performed
- What records must be completed
- What precautions must be taken to avoid contamination
If these points are not written clearly during drafting, the SOP may fail during review or cause operational errors later.
2. Review
After the draft is prepared, it moves to the review stage. Review is not a formality. It is a critical control point where the document is examined by different departments to ensure that it is correct, compliant, practical, and aligned with GMP requirements.
Pharmaceutical documentation is cross-functional by nature. A document prepared by one department often affects several others. For that reason, the review stage usually involves a cross-functional team.
Purpose of review
The review stage ensures:
- Technical accuracy
- GMP compliance
- Alignment with company policy
- Regulatory acceptability
- Practical usability in the actual working environment
Departments commonly involved in review
- Quality Assurance (QA) – checks compliance and document structure
- Production – confirms shop-floor practicality
- Quality Control (QC) – verifies laboratory relevance where needed
- Engineering – checks technical details for equipment-related documents
- Regulatory Affairs – ensures regulatory compatibility, especially for export plants
- Warehouse or Supply Chain – may review documents related to materials handling
What reviewers check
During review, reviewers may evaluate:
- Is the procedure correct and complete?
- Are responsibilities clearly assigned?
- Is there any ambiguity in wording?
- Are safety and contamination control measures included?
- Does the process match current operations?
- Are forms and records attached where required?
- Does the document conflict with any existing SOP or policy?
Why review matters
A document that is technically correct but operationally impractical can still fail. For example, an SOP may describe a sampling procedure that looks good on paper but does not match the actual equipment setup in the production area. The review stage helps detect and correct such issues before approval.
3. Approval
Once the review is completed and all comments are addressed, the document goes for approval. In the pharmaceutical industry, approval gives the document official status. Until approval is granted, the document remains a draft and cannot be used in GMP operations.
In most pharmaceutical organizations, the final approval authority rests with Quality Assurance (QA) because QA is the guardian of compliance, documentation control, and GMP systems.
Why approval is essential
Approval confirms that:
- The document has been reviewed properly
- All necessary corrections were made
- The content is compliant with GMP and company policy
- The document is suitable for operational use
Who approves documents?
Depending on document type, approval may involve:
- Department Head
- QA Manager
- Head of Quality
- Plant Head or authorized signatory for high-level documents
Approval of different document types
- SOPs are usually approved by QA and department heads
- Quality manuals may need senior management approval
- Validation protocols may require QA, engineering, and validation head approval
- Batch records and master documents often require QA authorization
Key point
A document without proper approval is considered uncontrolled and should never be used in the pharmaceutical environment.
4. Issuance
After approval, the document moves to the issuance stage. This is the point at which the document becomes an official controlled document and is released for use.
Issuance is one of the most important document control activities because it formally establishes the document’s identity, traceability, and status within the organization.
What happens during issuance?
- A unique document number is assigned
- Revision or version number is recorded
- Effective date is defined
- Department ownership is identified
- Controlled copy status is marked
- Master copy is secured by QA or document control
Why document numbering matters
A structured numbering system helps in:
- Easy retrieval
- Department-wise tracking
- Revision control
- Audit traceability
Example of a document number
A company may use a code like:
SOP-QA-015-V03
This may indicate:
- SOP = Standard Operating Procedure
- QA = Quality Assurance Department
- 015 = serial number
- V03 = version 3
Effective date
The effective date is important because it tells employees from which date the document becomes mandatory for implementation.
5. Distribution
Once issued, the document must be distributed in a controlled way. This stage is called distribution. In pharma, distribution does not mean simply sending a copy to everyone. It means ensuring that only authorized users have access to the approved and current version.
Objectives of controlled distribution
- Ensure availability of current documents where needed
- Prevent use of obsolete versions
- Limit access to authorized personnel only
- Maintain accountability for copies issued
Types of distribution
Controlled hard copies
These are printed copies with copy numbers, stamps, or signatures, distributed to specific departments.
Electronic access
In companies using EDMS, access is provided through authorized user accounts with role-based permissions.
Key controls during distribution
- Distribution list maintained
- Controlled copy log maintained
- Obsolete copies withdrawn
- Access restrictions applied where needed
Why unauthorized duplication is prohibited
If employees photocopy or unofficially share documents, outdated or altered versions may circulate, creating compliance risk and potential GMP failure. That is why pharmaceutical companies strictly prohibit unauthorized duplication of controlled documents.
6. Revision
A pharmaceutical document cannot remain unchanged forever. Processes evolve, regulatory requirements change, audit findings arise, and operational improvements are introduced. Therefore, documents must undergo revision whenever necessary.
Revision ensures that documents remain current, accurate, and aligned with actual practices.
Common reasons for revision
- Audit observations
- Process improvement
- Equipment modification
- Regulatory updates
- Product change
- Change control implementation
- CAPA requirements
- Error detected in existing document
What happens during revision?
- Revision need is identified
- Existing version is reviewed
- Changes are proposed
- Review and approval process is repeated
- New version is issued
- Previous version is marked obsolete
Importance of revision history
Every revised document should include a revision history section showing:
- Version number
- Date of revision
- Nature of change
- Approval details
This helps auditors and internal teams understand how the document has evolved over time.
7. Archival
The final stage of the lifecycle is archival. Once a document becomes obsolete or is replaced by a new version, it must not be discarded casually. It must be archived in a controlled manner according to company policy and regulatory requirements.
Archival preserves historical evidence and ensures traceability for inspections, product investigations, complaints, market recalls, and legal or regulatory review.
Documents commonly archived
- Obsolete SOPs
- Previous batch records
- Validation reports
- Training records
- Deviation files
- Change control files
- Audit reports
Retention period
Retention time depends on:
- Product type
- Market requirement
- Local regulation
- Company policy
In many cases, documents are retained for 5 to 10 years, but export-related documents or product-specific records may need longer retention.
Archival methods
- Controlled document archive room
- Fire-protected physical storage
- Secure digital servers
- Backup storage systems
Why archival matters
Archived documents are often required during:
- Regulatory inspections
- Product complaint investigations
- Trend analysis
- Legal defense
- Historical process review
🧾 Good Documentation Practices (GDocP) – Detailed Explanation
Good Documentation Practices, often called GDocP, are the rules that ensure pharmaceutical records are reliable, legible, traceable, and acceptable to regulators. In GMP-regulated environments, good documentation is not optional. It is a mandatory discipline.
Good Documentation Practices support data integrity, improve operational reliability, and provide evidence that activities were actually performed as required.
Why GDocP is so important
Pharmaceutical products directly affect patient health. Therefore, all manufacturing, testing, storage, release, and distribution activities must be properly documented. Poor documentation can create doubt about product quality, even if the actual process was performed correctly.
Regulators often apply a simple principle:
If it is not documented, it is considered not done.
Core rules of Good Documentation Practices
Write using indelible ink
When records are completed manually, entries should be made using permanent ink that cannot be easily erased or altered. Pencil is not acceptable because it can be changed without trace.
Do not use correction fluid
Correction fluid hides the original entry and destroys traceability. In pharma, original information must remain visible even if it was entered incorrectly.
Strike errors with a single line
If a mistake is made, the incorrect entry should be crossed out with a single line so that the original text remains readable.
Sign and date corrections
Any correction must include:
- Signature or initials
- Date of correction
- Reason, if required by SOP
Record data immediately
Data must be recorded at the time the activity is performed, not later from memory.
Write legibly
If records cannot be read clearly, they lose value and may be rejected during inspections.
Leave no blank spaces
Blank fields can be misused later. If a field does not apply, it should be marked as “N/A” according to procedure.
Use only approved formats
Employees must record data only on approved, current forms and templates.
Real GMP example
Wrong practice
An operator completes temperature readings at the end of the shift based on memory.
Correct practice
The operator records each temperature reading immediately at the actual time of observation.
Why the wrong practice is unacceptable
Writing data later introduces risk of:
- Memory error
- Backdating
- False records
- Data integrity breach
This is why regulators insist that data be contemporaneous and attributable.
🔁 Document Control Process Flow in a Pharmaceutical Company
A practical document control system in pharma consists of several sequential control steps. These steps ensure that documents are created, approved, implemented, reviewed, and retired in a systematic and GMP-compliant manner.
Step 1: User raises document request
A department identifies the need for a new document or revision. This may happen because of:
- New process introduction
- Audit observation
- Product transfer
- Regulatory requirement
- Change control
- Process improvement
The request is usually submitted to QA or document control.
Step 2: QA assigns document number
QA reviews the request and assigns a formal document number according to the company’s numbering system. This ensures traceability from the beginning.
Step 3: Draft created using template
The author prepares the document using an approved format. Templates are standardized so that all documents maintain consistency in structure and content.
Step 4: Review cycle initiated
The draft is circulated to relevant departments for technical, operational, and compliance review.
Step 5: QA approval
After review comments are incorporated, the document is approved by authorized personnel, usually with QA as the final authority.
Step 6: Document issued
The document is released as an official controlled document with version number and effective date.
Step 7: Employee training conducted
Before implementation, employees who will use the document must be trained. This is critical because a good SOP is ineffective if staff do not understand it.
Why training matters
Training ensures:
- Proper interpretation of procedure
- Consistent execution
- Reduction of human error
- Evidence of implementation
Step 8: Effectiveness check performed
Some companies perform an effectiveness check after implementation to confirm:
- Staff are following the document
- Procedure is practical
- No gaps remain
This may include supervisor observation, audit, or line walkthrough.
Step 9: Periodic review scheduled
Documents are not left untouched forever. They are reviewed periodically, often every 2 or 3 years, depending on company policy.
Step 10: Obsolete documents withdrawn
When a new version is issued, all old copies must be collected, stamped obsolete, archived, or removed from access points to prevent accidental use.
🇧🇩 Document Control in Bangladesh Pharmaceutical Industry
Document control has become increasingly important in the pharmaceutical industry of Bangladesh. As local companies expand into regulated export markets, documentation standards must improve to meet both domestic and international expectations.
Regulatory authority in Bangladesh
The pharmaceutical sector in Bangladesh is regulated by the Directorate General of Drug Administration (DGDA). DGDA oversees licensing, manufacturing standards, quality assurance expectations, and compliance with relevant drug regulations.
For companies targeting export markets, documentation systems often also need to align with:
- WHO GMP expectations
- Buyer requirements
- International regulatory expectations
- Customer audit standards
Why document control is becoming more important in Bangladesh
Bangladesh has a strong and growing pharmaceutical manufacturing base. Local companies are no longer focused only on domestic supply. Many are exporting products to international markets. This requires stronger systems for:
- Traceability
- Quality assurance
- Data integrity
- Inspection readiness
- Documentation control
As competition increases, companies with poor documentation systems will struggle to maintain credibility and compliance.
Industry leaders and current practices
Leading Bangladeshi pharmaceutical companies such as:
- Square Pharmaceuticals
- Beximco Pharmaceuticals
- Incepta Pharmaceuticals
are widely recognized for investing in better systems, stronger compliance structures, and more modern documentation approaches.
These companies have increasingly adopted:
- Electronic documentation systems
- Global compliance frameworks
- Strong QA oversight
- Audit-ready documentation practices
- Data integrity-focused controls
Their documentation maturity helps them compete in international markets and respond better to inspections, audits, and customer requirements.
⚙️ EDMS Features in Pharmaceutical Document Management
An Electronic Document Management System (EDMS) is a digital platform used to control pharmaceutical documents throughout their lifecycle. EDMS is becoming more important as companies move toward paperless or hybrid systems.
1. Version Control
Version control is one of the most essential EDMS features. It tracks the evolution of each document over time and ensures that only the current approved version is available for use.
Benefits of version control
- Prevents obsolete document use
- Maintains revision history
- Improves audit readiness
- Supports traceability of changes
2. Audit Trail
An audit trail automatically records actions taken within the system.
It typically records
- Who accessed the document
- Who modified the document
- What was changed
- Date and time of action
- Approval actions taken
Audit trails are crucial for data integrity and are highly valued during regulatory inspections.
3. Access Control
Not every employee should have the same level of access to documents. EDMS uses role-based permissions to control who can:
- View
- Edit
- Review
- Approve
- Archive
This reduces risk of unauthorized changes.
4. Electronic Signatures
Electronic signatures allow authorized users to approve and sign documents digitally. In compliant systems, electronic signatures are treated as legally meaningful and traceable.
Benefits
- Faster approvals
- Better traceability
- Reduced paper dependency
- Improved workflow efficiency
5. Workflow Automation
Workflow automation routes documents automatically through predefined steps such as drafting, review, approval, issuance, and training acknowledgment.
Benefits of workflow automation
- Faster turnaround time
- Fewer manual delays
- Better process visibility
- Reduced human error
- Easier status tracking
🔐 Data Integrity in Pharmaceutical Documentation
Data integrity means that data is complete, consistent, accurate, and reliable throughout its lifecycle. In pharmaceutical documentation, data integrity is not limited to laboratory results. It also includes SOP entries, batch records, logbooks, training records, and digital records.
Key requirements for data integrity
No unauthorized changes
Records must be protected from uncontrolled editing or manipulation.
Secure data storage
Both paper and electronic documents must be stored in a way that protects them from loss, damage, tampering, or unauthorized access.
Full traceability
Any action related to a record should be traceable to a person, time, and reason.
Regulatory expectations
Authorities expect companies to ensure:
- Real-time data entry
- No backdating
- No undocumented changes
- Complete audit trail
- Secure retention
- Reliable backups
ALCOA+ in document control
Data integrity is commonly summarized through ALCOA+ principles:
- Attributable
- Legible
- Contemporaneous
- Original
- Accurate
- Complete
- Consistent
- Enduring
- Available
A robust document control system must support all of these principles at every stage.
⚠️ Challenges in Bangladesh Pharmaceutical Documentation Systems
Despite industry growth, many Bangladeshi pharmaceutical companies still face practical challenges in document control.
1. Paper-based systems
Many smaller or mid-sized facilities still rely heavily on manual documentation.
Problems with paper systems
- Slow retrieval
- Risk of missing pages
- Limited traceability
- Greater storage burden
- Manual reconciliation challenges
2. Training gaps
Not all employees understand the importance of GMP documentation.
Common consequences
- Incomplete records
- Late entries
- Illegible handwriting
- Unapproved corrections
- Poor SOP compliance
3. Data integrity risks
Manual environments carry higher data integrity risk because they depend heavily on discipline and supervision.
Common risks include
- Backdated entries
- Missing signatures
- Unclear corrections
- Incomplete forms
- Untraceable changes
4. Cost constraints
Implementing a full EDMS, validating software, training staff, and maintaining system security requires investment. Smaller firms may delay adoption because of budget limitations.
However, the long-term cost of poor documentation can be much higher due to batch rejection, audit failure, rework, and reputational loss.
✅ Best Practices for Effective Document Control
A strong pharmaceutical document control system is built on disciplined practices, not just software.
1. Gradual EDMS implementation
Companies that cannot digitize everything at once can start with high-priority areas such as:
- SOP control
- Quality manuals
- Training records
- Controlled forms
Then they can gradually expand to:
- Batch records
- Validation files
- Change control
- Deviations and CAPA
2. Standardization
Uniform templates, numbering systems, review workflows, and correction practices reduce confusion and improve consistency across departments.
3. Continuous training
Documentation training should not be one-time only. Employees need:
- Induction training
- Refresher GMP training
- Practical documentation workshops
- Department-specific SOP training
4. Internal audits
Internal audits help identify documentation weaknesses before regulators or customers do.
Audits can detect
- Obsolete SOP use
- Missing signatures
- Delayed entries
- Incomplete records
- Poor archival practices
5. Strong version control
Version control is essential to prevent operational errors caused by outdated instructions. The company must ensure that only the latest approved document is in use everywhere.
Future Trends in Pharmaceutical Document Management
The future of document control in Bangladesh pharma will likely be shaped by digitalization and smarter quality systems.
1. Digital pharma
More paperless and hybrid systems will be introduced.
2. AI-assisted review
AI may help with document review support, metadata extraction, and searchability.
3. Blockchain potential
Though still emerging, blockchain may support tamper-evident records in some applications.
4. Cloud-enabled control
Secure cloud systems may support multi-site access, backup, and centralised control if properly validated.
💼 Career Opportunities in Document Control in Bangladesh Pharma
As pharmaceutical companies strengthen compliance systems, document control is becoming an increasingly important career area.
Common job roles
Document Controller
Responsible for issuance, tracking, filing, retrieval, and archival of controlled documents.
QA Executive
Handles SOP control, document review, training coordination, deviations, CAPA, and audit support.
Compliance Officer
Monitors GMP compliance, documentation discipline, and audit readiness.
Regulatory Affairs Specialist
Works with regulatory submissions, dossier documentation, product files, and compliance records.
Skills required
To build a strong career in pharmaceutical document control, candidates usually need:
- Knowledge of GMP and GDP
- Understanding of SOP structures
- Strong written communication
- Attention to detail
- Basic understanding of QA systems
- Familiarity with data integrity concepts
- Ability to manage records systematically
Conclusion
A strong Document Control & Management System in the pharmaceutical industry in Bangladesh is not optional. It is a core part of GMP compliance, data integrity, quality assurance, and business credibility. In a regulated environment, documents are not simply administrative tools. They are evidence, control mechanisms, and quality safeguards.
Companies that build disciplined document systems can improve:
- Regulatory readiness
- Product consistency
- Export competitiveness
- Investigation capability
- Audit performance
For pharmaceutical professionals, document control is also a valuable career pathway. Whether working in QA, QC, Production, Regulatory Affairs, or Compliance, strong documentation knowledge creates long-term professional advantage.
In simple terms, good documents support good decisions, good compliance, and good products.
FAQ: Document Control in Pharmaceutical Industry
What is document control in pharma?
Document control in pharma is the system used to create, review, approve, issue, revise, distribute, and archive documents so that only current approved versions are used.
Why is document control important in the pharmaceutical industry?
It supports GMP compliance, product quality, traceability, data integrity, and audit readiness.
What is the difference between DMS and EDMS in pharma?
A DMS may be manual or digital, while an EDMS specifically refers to an electronic system with digital workflows, audit trails, and automated controls.
What are the most important pharmaceutical documents?
Important documents include SOPs, Batch Manufacturing Records, Batch Packaging Records, validation documents, change controls, and deviation reports.
What is GDocP in pharma?
Good Documentation Practices are the rules that ensure records are complete, legible, accurate, timely, and traceable.
What jobs are available in document control in Bangladesh pharma?
Common roles include Document Controller, QA Executive, Compliance Officer, and Regulatory Affairs Specialist.