
Ensuring Data Integrity, Compliance & Patient Safety
🔍 Introduction to CSV in Pharma
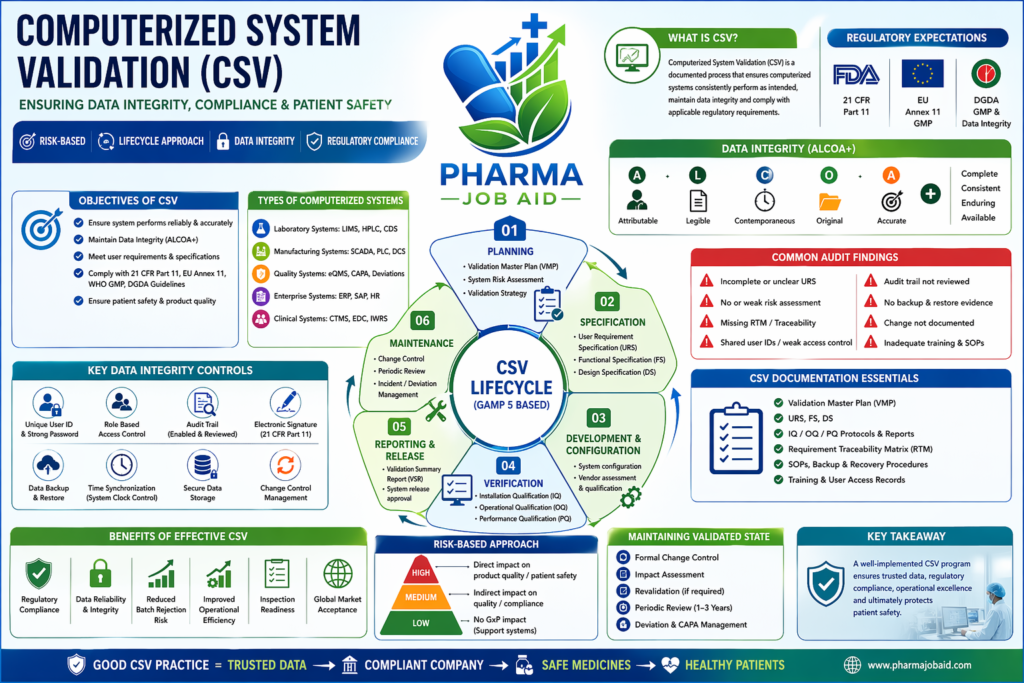
In today’s pharmaceutical manufacturing environment, computerized systems are deeply integrated into every stage of the product lifecycle—from research and development to manufacturing, quality control, and distribution. Computerized System Validation (CSV) is a documented process that ensures these systems consistently perform as intended, maintain data integrity, and comply with regulatory requirements such as US Food and Drug Administration, European Medicines Agency, and local authorities like the Directorate General of Drug Administration.
CSV is not just a regulatory requirement—it is a critical component of Quality Assurance (QA) that directly impacts product quality, patient safety, and company credibility.
🎯 Objectives of CSV
The primary objective of CSV is to provide documented evidence that a computerized system:
- Performs reliably and accurately
- Produces consistent and reproducible results
- Maintains data integrity (ALCOA+) principles
- Meets predefined specifications and user requirements
- Complies with 21 CFR Part 11 and EU Annex 11
🧩 Types of Computerized Systems in Pharma
Computerized systems requiring validation include:
- Laboratory Systems: LIMS, HPLC software, CDS
- Manufacturing Systems: SCADA, PLC, DCS
- Quality Systems: eQMS, deviation/CAPA software
- Enterprise Systems: ERP, SAP
- Clinical Systems: CTMS, EDC
Each system must be validated based on its GxP impact and risk level.
🔄 CSV Lifecycle Approach (GAMP 5 Based)
The CSV process follows a lifecycle approach aligned with International Society for Pharmaceutical Engineering GAMP 5 guidelines.
1. Planning Phase
- Validation Master Plan (VMP)
- System Risk Assessment (FMEA-based)
- Validation Strategy
2. Specification Phase
- User Requirement Specification (URS)
- Functional Specification (FS)
- Design Specification (DS)
3. Development & Configuration
- System configuration
- Vendor assessment & qualification
4. Verification Phase
- Installation Qualification (IQ)
- Operational Qualification (OQ)
- Performance Qualification (PQ)
5. Reporting & Release
- Validation Summary Report (VSR)
- System release approval
6. Maintenance Phase
- Change control
- Periodic review
- Incident/deviation management
⚙️ Key Validation Activities Explained
✅ Installation Qualification (IQ)
Ensures the system is installed correctly according to manufacturer specifications.
✅ Operational Qualification (OQ)
Verifies that the system operates as intended under defined conditions.
✅ Performance Qualification (PQ)
Confirms the system performs effectively in real-world operational conditions.
🔐 Data Integrity & CSV
One of the most critical aspects of CSV is ensuring data integrity. Regulatory agencies emphasize ALCOA+ principles:
- Attributable
- Legible
- Contemporaneous
- Original
- Accurate
(+ Complete, Consistent, Enduring, Available)
Failure in CSV often leads to data integrity violations, which are among the most common findings during DGDA and FDA inspections.
⚠️ Common Audit Findings (Bangladesh Pharma Context)
During DGDA inspections, the following CSV gaps are frequently observed:
- Incomplete URS or poorly defined requirements
- Lack of traceability matrix
- Missing or weak risk assessment
- No documented backup & recovery testing
- Inadequate access control (shared passwords)
- Absence of audit trail review procedures
📊 Risk-Based Approach in CSV
Modern CSV follows a risk-based approach, focusing validation effort where it matters most:
- High Risk: Direct impact on product quality (e.g., HPLC software)
- Medium Risk: Indirect impact (e.g., LIMS)
- Low Risk: No GxP impact (e.g., HR systems)
This approach is aligned with GAMP 5 Second Edition and reduces unnecessary documentation while maintaining compliance.
🔄 Change Control & Lifecycle Management
Once validated, systems must be maintained in a validated state through:
- Formal Change Control System
- Impact Assessment
- Revalidation (if required)
- Periodic Review (every 1–3 years as per SOP)
📑 Documentation in CSV (Audit-Ready)
Essential documents include:
- Validation Master Plan (VMP)
- User Requirement Specification (URS)
- Risk Assessment Report
- IQ/OQ/PQ Protocols & Reports
- Traceability Matrix (RTM)
- SOPs for system use and maintenance
- Backup & Disaster Recovery Procedures
Why CSV is Critical Today
With the rapid digitalization of pharmaceutical operations—ranging from automated manufacturing systems to cloud-based quality platforms—CSV has evolved from a documentation exercise into a risk-driven, lifecycle-based quality system. Regulatory agencies like the US Food and Drug Administration, European Medicines Agency, and Directorate General of Drug Administration now expect companies to demonstrate not only validation, but also continuous control over computerized systems.
In modern inspections, regulators focus less on volume of documentation and more on system understanding, risk control, and data integrity assurance.
Risk Assessment Methodology in CSV
A robust CSV program begins with a scientifically justified risk assessment, typically aligned with ICH Q9 Quality Risk Management principles. The goal is to identify which system functions directly impact product quality, patient safety, and data integrity.
🔬 Common Risk Assessment Tools
- FMEA (Failure Mode and Effects Analysis)
- HACCP (Hazard Analysis and Critical Control Points)
- FTA (Fault Tree Analysis)
📊 Example (Bangladesh Pharma Context)
For an HPLC software system:
- Risk Factor: Incorrect integration calculation
- Impact: Wrong assay result → batch rejection risk
- Severity: High
- Decision: Full OQ/PQ testing required
In contrast, for a document management system (DMS):
- Risk Factor: Incorrect document version control
- Impact: SOP non-compliance
- Severity: Medium
- Decision: Limited validation + strong procedural control
👉 This approach ensures regulatory focus on critical areas, which is strongly preferred during DGDA inspections.
🔗 Requirement Traceability Matrix (RTM)
The RTM is one of the most important audit documents in CSV. It ensures that:
- Every URS requirement is tested
- Every test case is justified
- No requirement is missed
📌 Typical RTM Structure
| URS ID | Requirement | Risk Level | Test Case | IQ/OQ/PQ Ref | Status |
|---|
👉 DGDA auditors often check RTM first to verify traceability and completeness.
🏭 Vendor Management & Supplier Qualification
Modern pharma companies rely heavily on third-party software vendors. Regulatory expectation is clear:
“You can outsource the system, but NOT the responsibility.”
🔍 Vendor Qualification Includes:
- Vendor audit (on-site or remote)
- Quality system evaluation
- Review of vendor documentation (IQ/OQ support)
- SLA (Service Level Agreement)
- Data security assurance
For example, ERP systems like SAP or cloud-based LIMS must be supported by vendor validation packages, but still require user-side verification.
☁️ Cloud Systems Validation (Modern Challenge)
Cloud-based systems are now widely used in pharma, but they introduce additional regulatory concerns.
⚠️ Key Risks:
- Data security & privacy
- Server location (cross-border data transfer)
- Access control
- Vendor dependency
✅ Regulatory Expectation:
Even for SaaS systems:
- Perform supplier assessment
- Define data ownership clearly
- Validate intended use (URS-based testing)
- Ensure backup and disaster recovery
👉 This is increasingly inspected by regulators globally.
🔐 Advanced Data Integrity Controls in CSV
Regulators emphasize data lifecycle control, not just system validation.
🔍 Critical Controls:
- Unique user ID & password policy
- Role-based access control
- Audit trail (enabled + reviewed periodically)
- Electronic signature compliance (aligned with 21 CFR Part 11)
- Data backup & restore verification
- Time synchronization (system clock control)
📌 Real Audit Observation (Common)
“Audit trail available but not reviewed”
👉 This is considered a major data integrity gap
🔄 Periodic Review (Maintaining Validated State)
Validation is not a one-time activity. Systems must remain in a validated state throughout lifecycle.
📅 Periodic Review Includes:
- Change history review
- Deviation/incidents review
- Access rights review
- Backup verification
- Performance monitoring
⏱️ Frequency:
- Typically 1–3 years (as per SOP and system criticality)
⚠️ Deviation & Incident Management in CSV
Any unexpected system behavior must be handled through a formal deviation system.
🔍 Examples:
- System crash during batch recording
- Data loss event
- Calculation error in software
- Unauthorized access
📌 Investigation Approach:
- Root Cause Analysis (5 Why / Fishbone)
- Impact assessment (Product/Data)
- CAPA implementation
- Revalidation (if needed)
📊 CSV Metrics for Management Review (MRM)
For DGDA-style Management Review Meeting (MRM), CSV performance should be tracked through KPIs:
- Number of validated systems
- % systems under change control
- Number of CSV deviations
- Audit trail review compliance rate
- Periodic review completion status
👉 These metrics demonstrate QMS maturity and digital compliance control.
🧾 SOP Structure for CSV (DGDA-Ready Format)
A standard CSV SOP should include:
- Purpose
- Scope
- Responsibility
- Definitions
- Validation Lifecycle Procedure
- Risk Assessment Method
- Documentation Requirement
- Change Control
- Periodic Review
- Deviation Handling
- Annexures (Templates)
👉 Must follow GDocP (Good Documentation Practice) and your internal SOP format (as per your company standard).
🧪 Real Case Study (Audit Scenario)
📍 Scenario:
During inspection, DGDA auditor reviews HPLC system.
❌ Findings:
- No documented OQ testing for integration calculation
- Shared user login used by analysts
- Audit trail enabled but not reviewed
🔍 Root Cause:
- Weak CSV implementation
- Lack of QA oversight
✅ CAPA:
- Revalidation (OQ + PQ)
- User access restructuring
- Monthly audit trail review SOP implemented
👉 This is a very common real-world situation in Bangladesh pharma audits.
📈 Integration with Other QMS Elements
CSV is not standalone—it is integrated with:
- Change Control System
- Deviation Management
- CAPA System
- Training Management
- Internal Audit Program
👉 During inspections, regulators assess how well CSV is linked with overall QMS.
🚀 Advanced Trend: Automation & AI in CSV
With increasing adoption of AI in pharma systems, regulators are cautious.
Recent cases (e.g., FDA warning letters) highlight:
- Over-reliance on AI without validation
- Lack of human oversight
- Poor understanding of system logic
👉 Key expectation:
AI systems must be validated like any other computerized system, with clear logic understanding and risk control.
🏁 Final Conclusion (Advanced Perspective)
Computerized System Validation (CSV) has transformed into a strategic quality function, essential for ensuring:
🔍 Inspection Readiness Strategy for CSV (DGDA / FDA / EU)
Regulatory inspections today are highly data-integrity driven. Inspectors from the Directorate General of Drug Administration, US Food and Drug Administration, and European Medicines Agency follow a system-traceability approach, meaning:
They start from a data point → trace back to system → then to validation evidence
🧭 Typical Inspection Flow:
- Select a batch record / lab result
- Identify the computerized system used
- Request validation documents (URS, IQ/OQ/PQ)
- Verify RTM linkage
- Check audit trail and access control
- Review change history
- Evaluate data integrity compliance
👉 If any link is weak → Major observation raised
🧾 Audit-Ready CSV Document Package (Complete List)
To be fully inspection-ready, each GxP computerized system should have:
📁 Core Documents:
- Validation Master Plan (VMP)
- User Requirement Specification (URS)
- Functional Specification (FS)
- Design Specification (DS)
- Risk Assessment Report
🧪 Qualification Documents:
- IQ Protocol & Report
- OQ Protocol & Report
- PQ Protocol & Report
🔗 Control Documents:
- Requirement Traceability Matrix (RTM)
- SOPs (Operation, Backup, Security, Audit Trail Review)
- Change Control Records
- Deviation / Incident Reports
🔐 Data Integrity Documents:
- User Access Matrix
- Audit Trail Review Records
- Backup & Restore Test Evidence
👉 Missing even one critical document can trigger a regulatory finding.
📊 CSV Checklist (DGDA Audit Quick Tool)
Here is a practical audit checklist you can directly use:
| Checkpoint | Requirement | Status |
|---|---|---|
| URS Approved | QA approved, clear requirements | ☐ |
| Risk Assessment | Documented & justified | ☐ |
| IQ Completed | Installation verified | ☐ |
| OQ Completed | Functional testing done | ☐ |
| PQ Completed | Real-use verification | ☐ |
| RTM Available | Full traceability | ☐ |
| Audit Trail Enabled | System configured | ☐ |
| Audit Trail Reviewed | Periodic review done | ☐ |
| Access Control | Unique user ID | ☐ |
| Backup Tested | Restore verified | ☐ |
| Change Control | All changes documented | ☐ |
👉 This checklist is extremely useful for self-inspection before DGDA audit.
🏭 Department-Wise Responsibility in CSV
CSV is a cross-functional activity, not only QA responsibility.
👨🔬 Quality Assurance (QA)
- Approve validation documents
- Ensure compliance
- Conduct periodic review
💻 IT Department
- System installation & maintenance
- Backup & security management
- Technical support
🏭 User Department (QC/Production)
- Define URS
- Execute PQ
- Report deviations
🧑💼 Management
- Approve validation strategy
- Provide resources
- Review CSV metrics (MRM)
👉 Weak coordination between these departments is a common audit gap.
⚠️ Top Critical Deficiencies Observed in Pharma Audits
Based on real inspection trends:
🔴 Critical Findings:
- No validation for critical systems (e.g., HPLC software)
- Data deletion possible without audit trail
- Shared login IDs
- No backup or failed recovery test
🟠 Major Findings:
- Incomplete URS
- Missing RTM
- Audit trail not reviewed
- No periodic review
🟡 Minor Findings:
- Formatting issues in documents
- Missing signatures/dates
👉 Data integrity findings are always treated as critical risk
🧪 Practical CSV Implementation Roadmap (Step-by-Step)
For a new pharmaceutical company or system:
🚀 Step 1: System Identification
List all computerized systems (GxP vs non-GxP)
🚀 Step 2: Risk Categorization
High / Medium / Low impact classification
🚀 Step 3: Prepare URS
Clear, testable requirements
🚀 Step 4: Perform Validation
Execute IQ/OQ/PQ based on risk
🚀 Step 5: Documentation
Prepare RTM, reports, SOPs
🚀 Step 6: Release System
QA approval before use
🚀 Step 7: Lifecycle Management
Change control + periodic review
👉 This roadmap aligns perfectly with GAMP 5 lifecycle model.
🔄 Revalidation: When Is It Required?
Revalidation is necessary when:
- Major system upgrade
- Software version change
- Hardware replacement
- Process change affecting system use
- Regulatory requirement
📌 Example:
Upgrading HPLC software version → requires partial or full OQ re-execution
🧠 Human Factor in CSV (Most Ignored Risk)
Even with a validated system, human error remains a major risk.
⚠️ Common Issues:
- Password sharing
- Ignoring audit trail review
- Incorrect data entry
- Bypassing SOP
✅ Control Measures:
- Regular training
- Access restriction
- SOP enforcement
- Internal audits
👉 Regulators strongly evaluate user behavior during inspection.
📈 Digital Transformation & CSV Maturity Model
Pharma companies can be categorized based on CSV maturity:
Level 1: Basic
- Paper-based validation
- Minimal risk assessment
Level 2: Structured
- SOP-driven validation
- Standard templates
Level 3: Advanced
- Risk-based validation
- Integrated QMS
Level 4: Optimized
- Automated validation tracking
- Real-time compliance monitoring
👉 Most Bangladesh pharma companies are currently at Level 2–3
🔗 CSV & Global Compliance
For exporting to regulated markets:
- USA → Compliance with 21 CFR Part 11
- EU → Compliance with EU Annex 11
- WHO → Data integrity guidelines
👉 CSV is a mandatory requirement for international market approval
🏭 CSV in Bangladesh Pharmaceutical Industry
In Bangladesh, CSV is increasingly emphasized by DGDA due to:
- Export requirements (EU, US markets)
- Data integrity concerns
- Digital transformation of pharma operations
Companies are now implementing eQMS, LIMS, and ERP systems, making CSV a critical compliance area.
🚀 Benefits of Effective CSV
- Ensures regulatory compliance
- Improves data reliability
- Reduces batch rejection risk
- Enhances inspection readiness
- Builds global market trust
❌ Risks of Poor CSV Implementation
- Regulatory warning letters
- Product recalls
- Data manipulation risks
- Loss of market authorization
- Reputational damage
📈 Future Trends in CSV
- Shift toward Computer Software Assurance (CSA) (FDA approach)
- Increased use of cloud-based systems
- Integration of AI/ML in pharma systems (with strict validation control)
- Automation of validation documentation
🧠 Conclusion
Computerized System Validation (CSV) is no longer optional—it is a regulatory necessity and business-critical function in the pharmaceutical industry. A well-implemented CSV framework ensures that computerized systems operate reliably, maintain data integrity, and support compliance with global regulatory standards.
For Bangladesh pharmaceutical companies aiming for international markets, robust CSV practices aligned with GAMP 5 and DGDA expectations are essential for sustainable growth and regulatory success.